Single-cell RNA sequencing data analysis

Single-cell RNA sequencing enables cataloging and studying cellular identities at a scale and resolution unmatched by bulk sequencing.
Single-cell RNA sequencing (scRNA-seq) is one of the most rapidly advancing and diversifying technologies in molecular biology. The ability to study gene expression on the resolution of single cells has been as transformative as the advent of bulk RNA-sequencing previously.
In addition to single-cell RNA-seq, a number of other next-generation sequencing (NGS) -based assays have been adapted to single-cell protocols. These include genomic, proteomic and epigenetic assays, notably single-cell ATAC-sequencing, which is commonly performed in conjunction with scRNA-seq.
Platforms and protocols for scRNA-seq vary in their throughput (number of cells) and transcript coverage (3'/5' tag-based vs whole-transcript). Our team has experience working with several technologies, such as 10X Genomics, Drop-Seq, BD Rhapsody system and protocols of the CEL-Seq and Smart-Seq families.
Here we present typical single-cell analyses, focusing on scRNA-seq but covering also its integration with other common single-cell assays.
Quality control and preprocessing
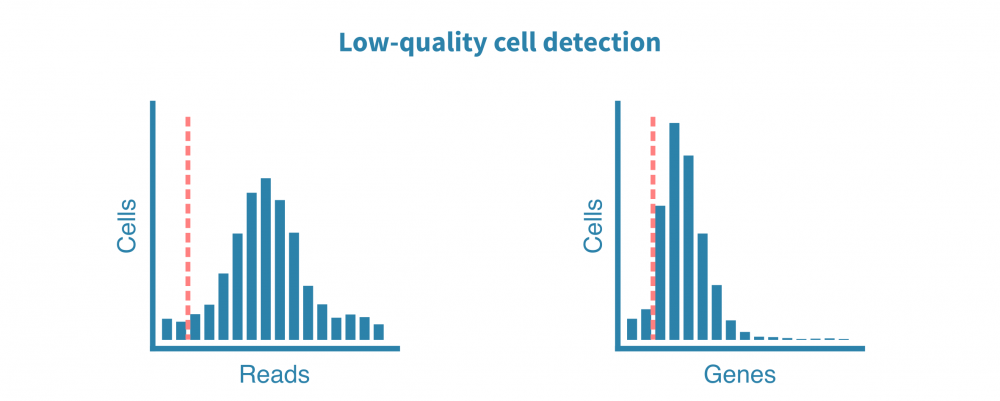
Like with any NGS data, the analysis of single-cell sequencing data starts with quality control and preprocessing.
Raw sequencing reads are quality-tested and metrics such as cell quality, accuracy, and diversity are generated. Reads are then aligned to an applicable reference genome or transcriptome, and additional metrics such as the number of cells, reads per cell, genes per cell, sequencing saturation and fraction of mitochondrial transcripts are plotted and inspected.
These QC metrics inform us about the total quality of the libraries and the usability of the samples and enable identifying and removing low-quality cells.
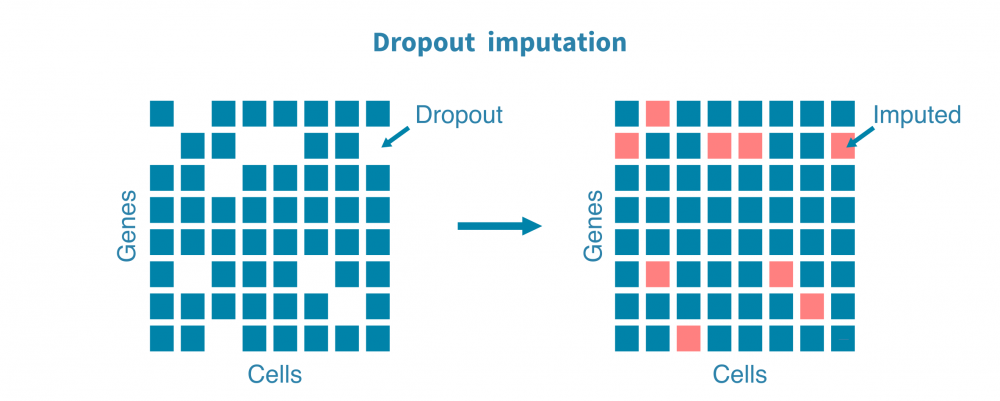
Further preprocessing is often carried out to remove unwanted signal, or noise, from certain downstream analyses. These include
- imputation to estimate read counts for dropouts, or genes with zero transcripts due to technical, rather than biological, reasons,
- normalization to remove biases due to e.g., differences in cell sizes, and
- reducing the data to representative variables such as highly-variable genes or principal components.
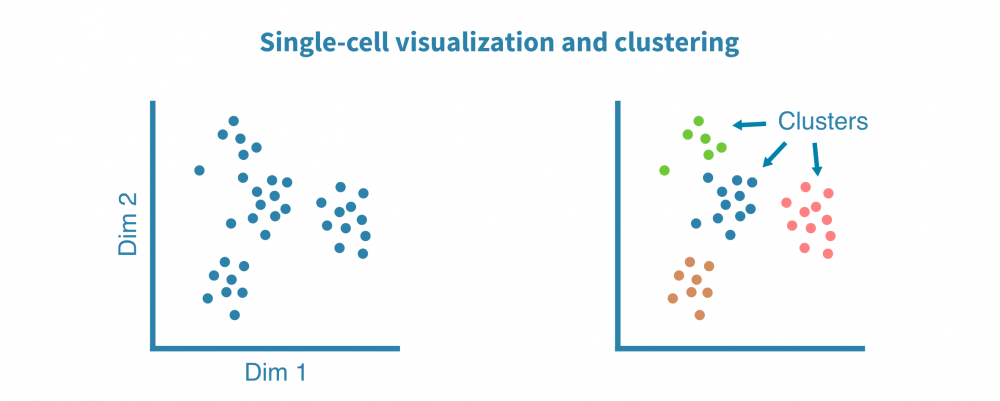
Exploratory analysis
Preprocessed single-cell RNA-seq data is clustered to identify groups of similar cells and visualized using non-linear dimensionality reduction algorithms such tSNE and UMAP and correlation heatmaps to unveil general patterns of cell heterogeneity.
These visualizations help us answer technical questions such as:
- Do the biological replicates resemble each other?
- Are there outlier samples or cells?
- Are the cell clusters distinct?
...and biological questions such as:
- How heterogeneous are the underlying cell types/states?
- Do distinct samples (e.g., different tissues, treatments or time points) form separate clusters?
Cell type identification
Identifying and characterizing cell types (and more refined cell states) is the most central part of most single-cell projects.
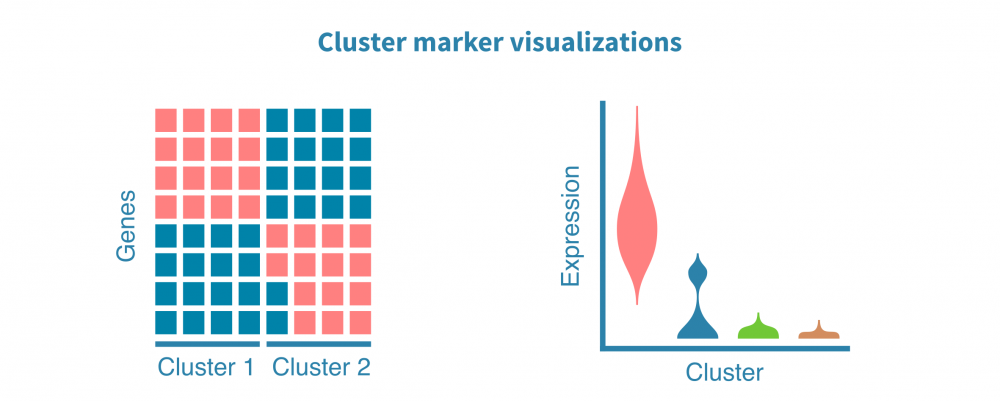
It all starts with identifying features (e.g., genes, proteins, accessible regions) that are specific to each cell cluster. These markers are defined by differential expression (DE) comparison of each cell cluster and the remaining ones, yielding DE statistics such as fold change and statistical significance.
The cluster markers can be visualized using scatter plots, violin plots, and heatmaps.
Markers are further annotated to biologically meaningful terms, such as a biological processes, signaling pathways or a specific disease. Such analyses may rely either on over-representation analysis or gene set enrichment analysis, which both result in a list of enriched gene sets with relevant statistics and annotations.
Single-cell datasets are typically also integrated with publicly available data in order to exploit the cell-type information from already annotated datasets or cell atlases. This enables transferring cell labels into the analyzed dataset.
The transferred cell labels and identified markers and their annotations are used, together with prior information on cell-type/state markers, to identify the captured cell types.

Trajectory analysis
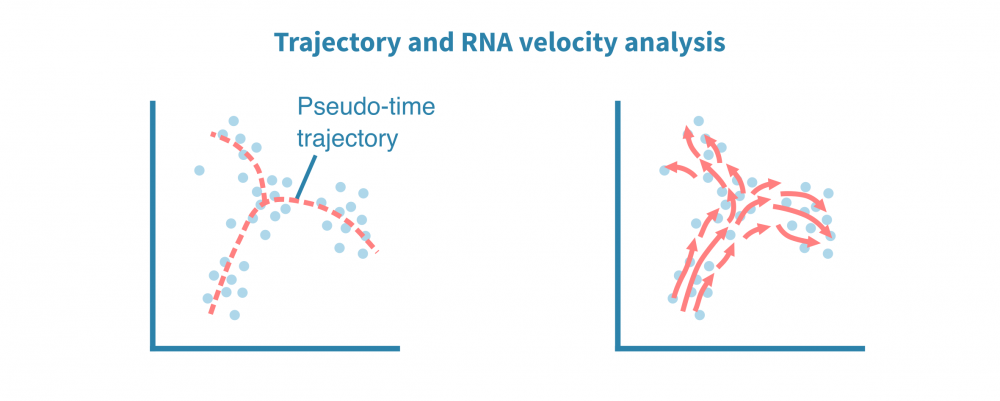
In addition to characterizing distinct cellular identities, single-cell data lends itself to identifying continuums of gradual change in cell state, or trajectories. Uncovering such continuums is also called pseudotime analysis — while all cells are sampled at the same time point, individual cells may represent different stages in a temporal process such as differentiation.
De novo reconstruction of lineage differentiation and cell maturation trajectories allow exploring cellular dynamics, delineation of cell developmental lineages, and characterization of transition between cell states along a latent pseudotime dimension.
An ensemble of trajectory inference algorithms may be used for robust identification of root and terminal cellular states, branching points, and lineages. Single cells are ranked across deterministic or probabilistic lineages, and their ranking indicates their progression in a dynamic process of interest.
This type of analysis may also utilize the ratio of processed and unprocessed transcripts to infer whether a gene's expression is increasing or decreasing in a given cell. Combining this information from all quantified genes at a given state enables inferring the direction and pace of change in states. This is called RNA velocity analysis.
Integrative single-cell analyses
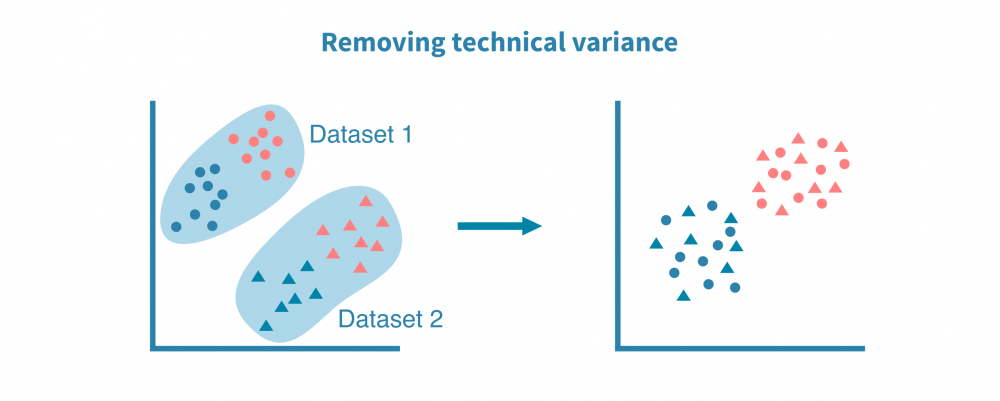
Integrative single-cell analyses bring different datasets, including different data types and species together. This enables more accurate and detailed cell labeling and mechanistic insight into gene regulation in the studied system. Such analyses rely on common properties, or anchors, between the datasets, such as matched features (e.g., genes or homologues) or matched cells.
Integrating multiple single-cell RNA-seq datasets
Perhaps the most common integration of single-cell datasets takes place between scRNA-seq datasets from different sources or technology platforms. Using genes as anchors, a successful integration removes the technical bias while retaining biological variance of the datasets.
Combining different scRNA-seq datasets is particularly helpful when there is a well-characterized public expression atlas available for a relevant tissue or organism.
Integrating single-cell RNA-seq and epigenomics
Integrating single-cell RNA-seq data with single-cell ATAC-seq or single-cell methylation data often relies on matched cells as anchors (when the measurements derive from the same cells as in, e.g., 10X Genomics Multiome technology).
Combining expression data with chromatin accessibility or methylation profiles enables more robust identification of cell types and allows for quantifying the effect of chromatin state to expression in individual cell types.
Read more about integrating epigenomics and transcriptomics
Integrating single-cell RNA-seq and proteomics
Since proteins, rather than transcripts, are key drivers of cellular functions, single-cell proteomics complements scRNA-seq experiments with more accurate estimates of cells functional states.
Single-cell proteomic profiling (CITE-seq, flow cytometry, mass cytometry, and mass spectrometry) comes in different degrees of throughput (number of proteins quantified) and can be targeted specifically to surface proteins, as in CITE-seq which involves a panel surface proteins quantified from cells with matched scRNA-seq reads.
Surface proteins are particularly useful in cell type identification, while the inclusion of cytosolic proteins enable better characterization of pathway and gene-regulatory activities.
Cross-species integration
Cross-species integrative analysis enables the identification of cell-type phylogenies that define the relationships of evolutionary and developmental mechanisms between different organisms. Shared homologues are used as anchors in cross-species integration.
This is particularly helpful when a disease/organ is better characterized on a single-cell resolution in an animal model than in human.
Ligand-receptor analysis
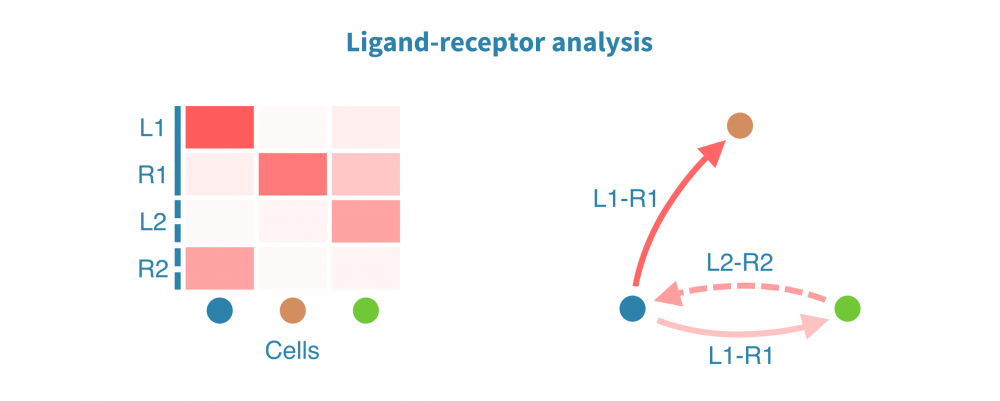
Ligand-receptor (LR) analysis uncovers cell-cell interactions that coordinate homeostasis, development, and other system-level functions. Changes and dysfunction in such interactions may go unnoticed in an analysis limited to the internal state of individual cells or cell types.
Ligand-receptor analysis identifies and quantifies intercellular interactions based on the expression of known receptors and their ligands. The interactions may take place within or between tissues, and the strength of this interaction is compared between biological conditions of interest, such as patient groups, disease states, and treatments.
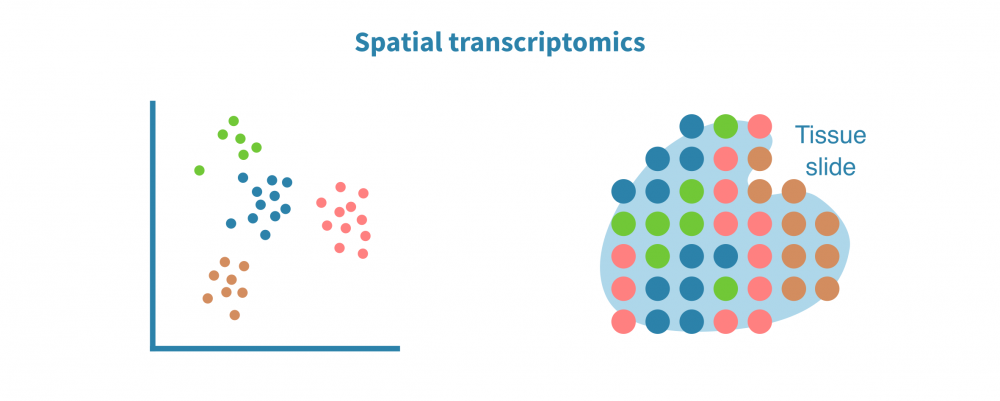
Spatial transcriptomic analysis
Spatially resolved single-cell transcriptomic assays couple expression data with the cell's positional context in a tissue or organ. This is particularly useful in the study of complex solid tissues, such as tumors and their microenvironment.
Spatial transcriptomic analysis involves cell/spot clustering in space, identification of spatially variable genes and resolving cell types in space.
Retaining the positional information of sequenced cells adds to the accuracy of identifying cell types and ligand-receptor interactions. It also enables spatial visualization of gene expression or chromatin accessibility (in the case of scATAC-seq) and integrating imaging-based data to the analysis.
Even in the case of lower-resolution assays, like 10X Visium, multimodal spatial analysis helps in correcting gene expression values and imputing dropout events.