单细胞RNA测序数据分析

单细胞RNA测序使得细胞鉴定和研究在规模和分辨率上达到了高于批量测序的水平。
单细胞RNA测序(scRNA-seq)是分子生物学中发展和多样化最快的技术之一。研究基因表达在单个细胞水平上的能力就像之前批量RNA测序的出现一样具有变革性。
除了单细胞RNA测序,还有许多其他基于下一代测序(NGS)的检测方法已被适应于单细胞协议。这些包括基因组学、蛋白质组学和表观遗传学检测,特别是单细胞ATAC测序,通常与scRNA-seq一起进行。
平台和scRNA-seq协议在其吞吐量(细胞数)和转录本覆盖率(3'/5'标签基础 vs 全转录本)方面有所不同。我们团队在多种技术方面具有经验,如10X Genomics、Drop-Seq、BD Rhapsody系统以及CEL-Seq和Smart-Seq系列的协议。
这里我们介绍典型的单细胞分析,重点是scRNA-seq,但也涵盖了其与其他常见的单细胞检测的整合。
质量控制和预处理
与任何NGS数据一样,对单细胞测序数据的分析始于质量控制和预处理。
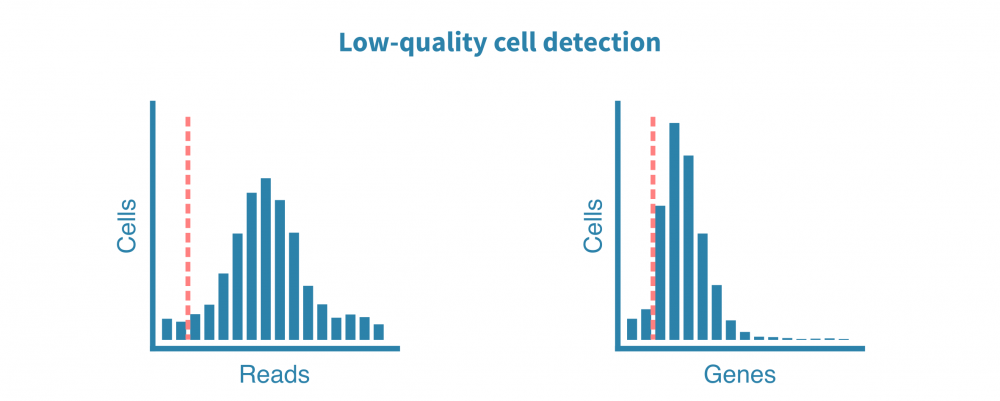
原始测序读数经过质量测试,并生成诸如细胞质量、准确性和多样性等指标。然后将读数与适当的参考基因组或转录组进行比对,并绘制和检查附加的指标,例如细胞数、每个细胞的读数、每个细胞的基因数、测序饱和度以及线粒体转录本的比例。
这些质控指标告诉我们关于文库的总体质量和样品的可用性,并使我们能够确定和去除低质量的细胞。
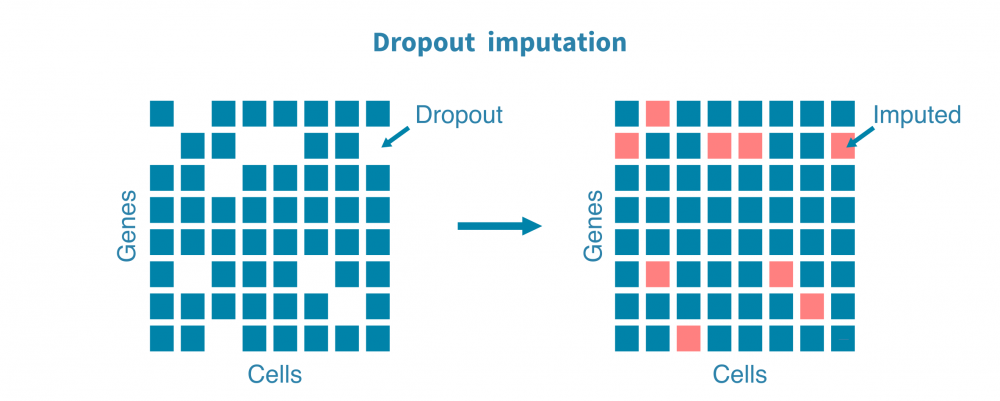
通常还进行进一步的预处理,以从某些下游分析中去除不需要的信号或噪声,这包括:
- 估计替代值以估计由于技术而非生物学原因而导致的漏读或零转录本的基因;
- 规范化以消除例如细胞大小差异等因素引起的偏差;以及
- 将数据降至代表性变量,如高变量基因或主成分。
探索性分析
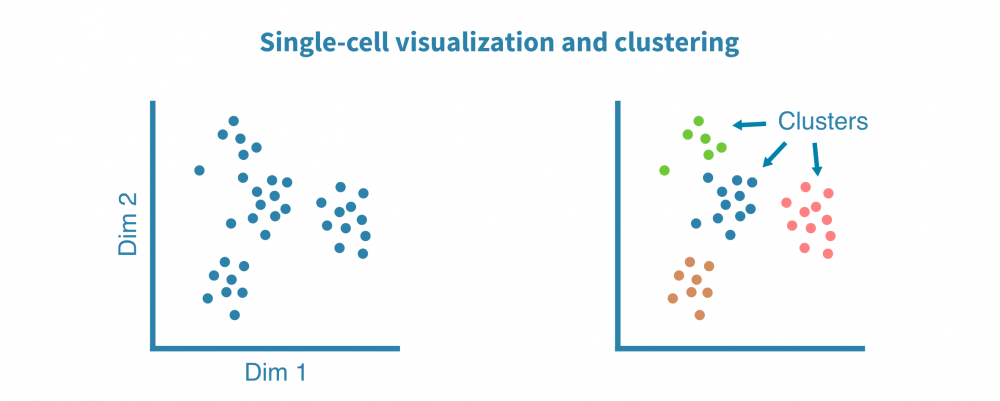
预处理的单细胞RNA测序数据被聚类以识别相似的细胞群,并使用非线性降维算法(如tSNE和UMAP)和相关性热图进行可视化,以揭示细胞异质性的一般模式。
这些可视化帮助我们回答技术问题,例如:
- 生物学重复是否相似?
- 是否有离群样本或细胞?
- 细胞群是否不同?
……以及生物学问题,例如:
- 基础细胞类型/状态有多么异质?
- 不同样本(例如不同组织、治疗或时间点)是否形成单独的群集?
细胞类型鉴定
识别和表征细胞类型(以及更精细的细胞状态)是大多数单细胞项目最核心的部分。
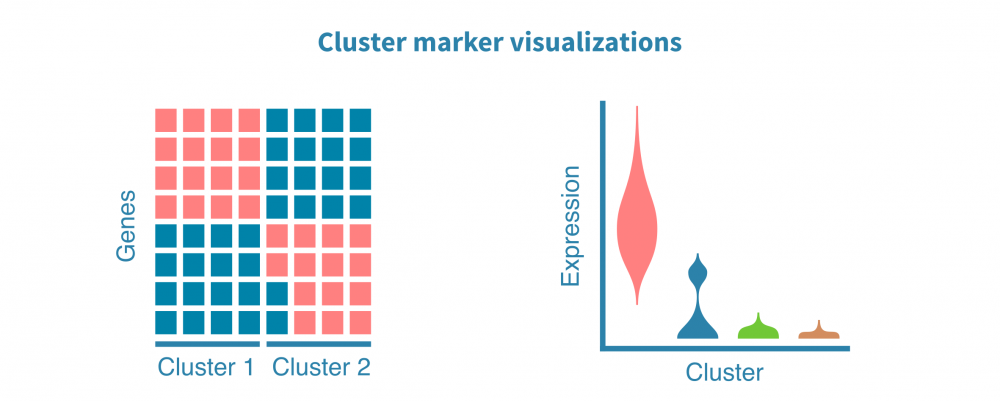
这一切始于识别特定于每个细胞群的特征(例如基因、蛋白质、可访问区域)。这些标记由差异表达(DE)比较每个细胞群和其余细胞群而定义,产生如折叠变化和统计显着性等DE统计量。
可以使用散点图、小提琴图和热图可视化细胞群标记。
标记进一步注释为生物学意义的术语,例如生物过程、信号通路或特定疾病。这些分析可能依赖于超表达分析或基因集富集分析,两者都会产生一系列富集的基因集与相关统计信息和注释。
单细胞数据集通常也与公共可用数据集集成,以利用已注释数据集或细胞图谱中的细胞类型信息。这使得将细胞标签转移至分析的数据集成为可能。
转移的细胞标签和鉴定的标记及其注释与关于细胞类型/状态标记的先前信息一起用于鉴定捕获的细胞类型。

轨迹分析
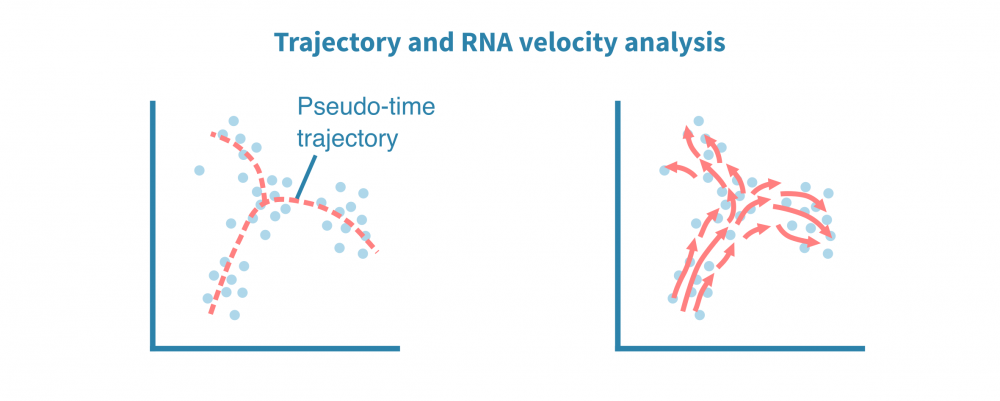
除了表征不同的细胞身份外,单细胞数据还适用于识别细胞状态渐变的连续体或轨迹。揭示这种连续体也被称为假时间分析,尽管所有细胞在同一时间点被采样,但个体细胞可能代表分化等时间过程中不同的阶段。
利用分化分支和细胞成熟轨迹的全新重建,可以探索细胞动态,勾勒细胞发育谱系,并表征沿着潜在假时间维度的细胞状态转换。
轨迹推断算法的集合可用于鲁棒地识别根和终端细胞状态、分支点和谱系。单细胞沿着确定性或概率谱系进行排序,它们的排序指示了它们在感兴趣的动态过程中的进展情况。
这种类型的分析还可以利用加工和未加工转录本的比率推断基因表达在给定细胞中是增加还是减少。将来自给定状态下所有定量基因的这些信息相结合,可以推断状态的变化方向和速度。这称为RNA速度分析。
综合单细胞分析
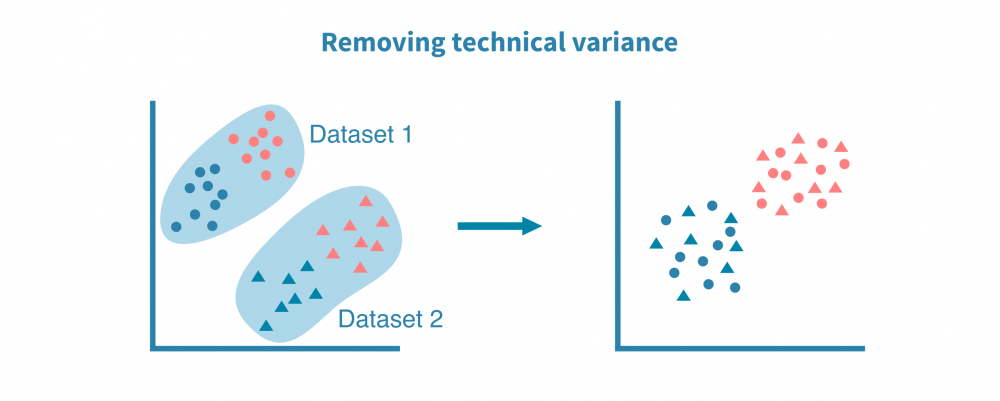
综合单细胞分析将不同的数据集,包括不同的数据类型和物种集成在一起,这使得对所研究系统中基因调控的机制有更准确和详细的细胞标记和洞察。这种分析依赖于数据集之间的共同属性或“锚点”,如匹配的特征(例如基因或同源物)或匹配的细胞。
整合多个单细胞RNA测序数据集
最常见的单细胞数据集整合是来自不同来源或技术平台的scRNA-seq数据集之间的整合。使用基因作为锚点,成功的整合可以去除数据集的技术偏差同时保留生物变异。
当有关于相应组织或生物体的公共表达图谱时,整合不同的scRNA-seq数据集特别有帮助。
整合单细胞RNA测序和表观遗传学
将单细胞RNA测序数据与单细胞ATAC-seq或单细胞甲基化数据结合起来通常依赖于匹配的细胞作为锚点(当测量来源于与10X Genomics Multiome技术中相同的细胞时)。
将表达数据与染色质可及性或甲基化数据相结合,可以更可靠地识别细胞类型,并允许量化染色质状态对各个细胞类型的表达的影响。
整合单细胞RNA测序和蛋白质组学
由于蛋白质而不是转录本是细胞功能的关键驱动因素,单细胞蛋白质组学通过更准确地估计细胞的功能状态来补充scRNA-seq实验。
单细胞蛋白质组学分析(CITE-seq,流式细胞术,质谱和质谱分析)具有不同的吞吐量(量化的蛋白质数量)并可以专门针对表面蛋白进行定向,如CITE-seq,它涉及从具有匹配scRNA-seq读数的细胞中量化表面蛋白。
表面蛋白在细胞类型鉴定中特别有用,而包含胞质蛋白则可以更好地表征通路和基因调控活动。
跨物种整合分析
跨物种综合分析可确定定义不同生物之间进化和发育机制关系的细胞类型谱系。在跨物种整合中,使用共享的同源物作为锚点。
当疾病/器官在动物模型中的单细胞分辨率上得到更好的表征时,这特别有助于人类疾病/器官的研究。
膜受体配体分析
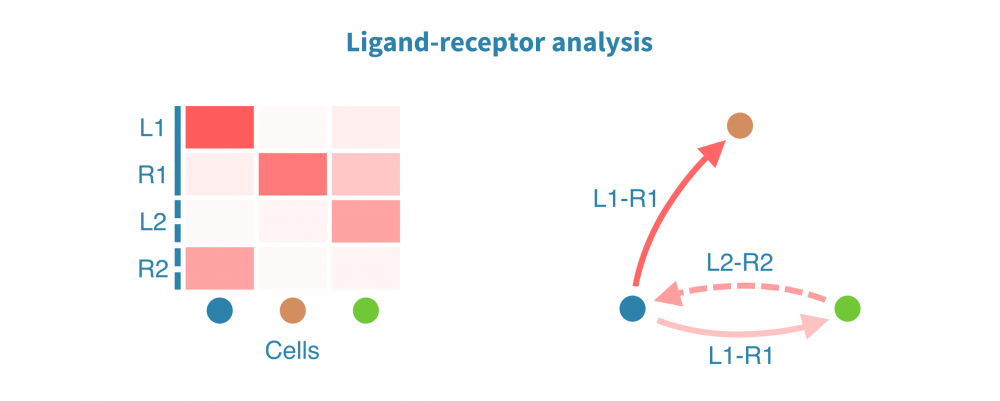
膜受体配体(LR)分析揭示了协调体内稳态、发育和其他系统级功能的细胞间相互作用。此类相互作用的变化和功能失调在仅限于个体细胞或细胞类型内部状态分析中可能不被注意到。
膜受体配体分析根据已知受体和其配体的表达量识别和量化细胞间相互作用。这些相互作用可能在组织内或组织间发生,其强度将在感兴趣的生物条件(如患者组、疾病状态和治疗)之间进行比较。
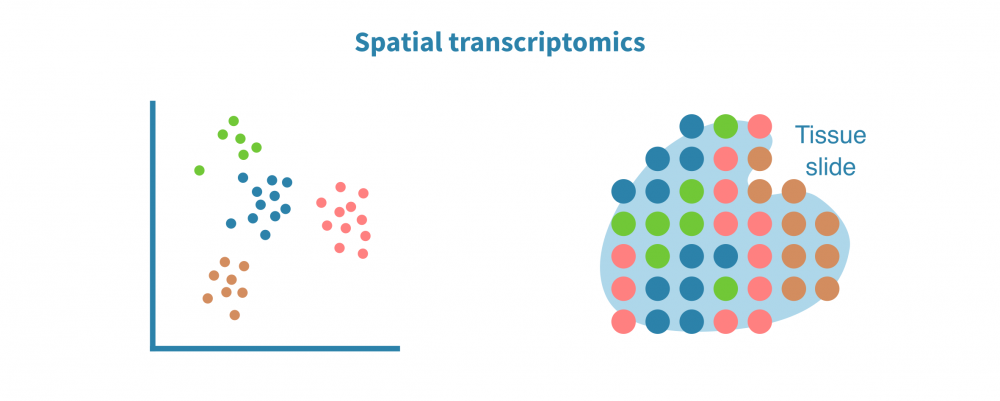
空间转录组分析
空间解析单细胞转录组学分析将表达数据与细胞在组织或器官中的位置上下文联系起来。这在研究肿瘤及其微环境等复杂实体组织中特别有用。
空间转录组分析包括空间中的细胞/点聚类、空间变量基因的识别和空间中的细胞类型解析。
保留序列化细胞的位置信息有助于准确识别细胞类型和膜受体配体相互作用。它还能够实现基因表达或染色质可及性(在scATAC-seq情况下)的空间可视化,并将基于成像的数据整合到分析中。
即使在像10X Visium这样的低分辨率分析中,多模式空间分析也有助于纠正基因表达值和补充数据缺失事件。