RNA-seq和ChIP-seq数据的整合分析
“你如何整合RNA-seq和ChIP-seq数据?”
我听到过许多次生物学家问这个问题(您可以将ChIP-seq替换为ATAC-seq、bisulphite-seq或任何其他 表观基因组数据 类型)。
这个问题很有意思。
生成和分析单个基于NGS的测序(如RNA-seq或ChIP-seq)的数据不像几年前那样罕见。这归功于新一代“NGS原生态”生物学家,他们在培训的早期就掌握了基本的“组学”数据分析技能,这在很大程度上消除了生物学家前往统计学系或计算机科学系的必要,向擅长编码的研究人员敲门,建议“合作”来分析他们的数据。
但是,整合不同的数据模式是另一回事,这是研究项目常常停滞的阶段。
这个想法很简单:如果您闻到、品尝和尝试一款葡萄酒,您的大脑可以整合这些多感官输入并推断出葡萄的产地,比如果只依赖一个感官更好。
那么,什么是可以采用您生成的所有可能的NGS数据并得出有洞见的多组学大脑?
我已经学会了错误答案:“这真的取决于您的研究问题。” 正确的答案是“相关性”。那是简短的答案——长的答案是“仔细分析各个数据类型,相关性,过滤,可视化,解释——迭代几次——你可能会得到一些非常好的结果!”
工作流程
为了介绍这种数据整合方法,让我们假设下面可视化的实验,在几个时间点进行了RNA测序和表观基因组测序实验,以及在第一个时间点后施加了一种处理。表观基因组测序实验可以是针对一种或多种组蛋白修饰的ChIP-seq(或CUT&Tag)实验,也可以是ATAC-seq等染色质可及性实验。
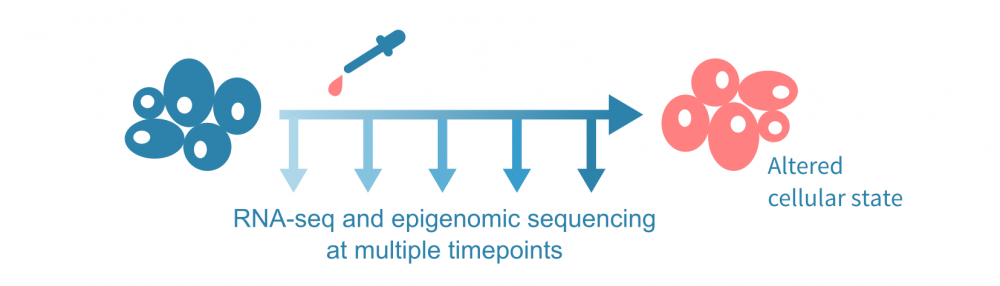
(这里讨论的综合分析不需要时间序列数据;可以使用 单细胞数据 沿伪时间轨迹分析表达和表观状态,或者仅比较来自不同条件的批量实验的单一时间点数据。)
问题是,在治疗和最终改变的细胞状态之间有哪些分子机制?我们能否给出一个多步骤的描述,通过基因和基因产物网络级联的事件来重新编程细胞以适应干扰?
在转化研究的背景下,识别关键元素,如转录因子或增强子,这些元素使得细胞进入疾病状态,可以为新疗法提供可能的靶点。
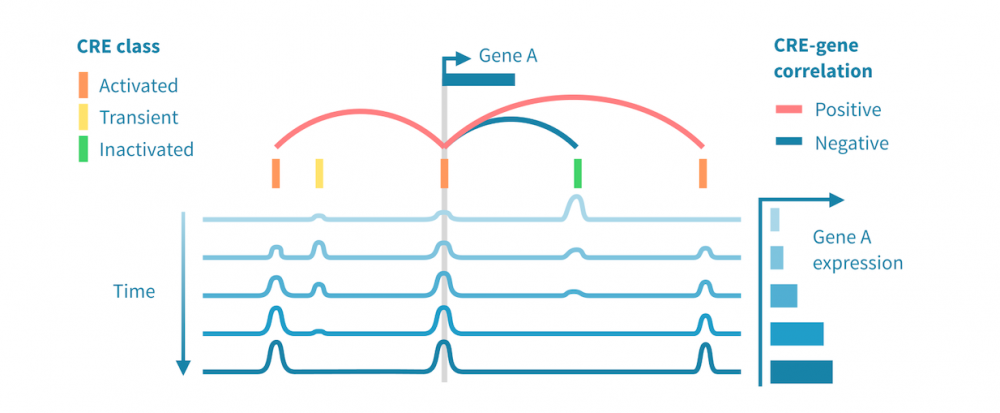
下面我们看到一个识别此类级联中活跃的顺式和反式调节路径的工作流程。它从分别处理表观基因组和转录组数据开始,并通过将每个基因的表达与其假定的顺式调节元素 (CREs) 的信号相关联,将这两种模态结合起来。然后继续通过识别驱动染色质变化的转录因子 (TFs),通过在 CREs 中识别 TF 结合基序并将 TF 的表达与这些假定的结合位点的状态相关联来确定这些 TFs。
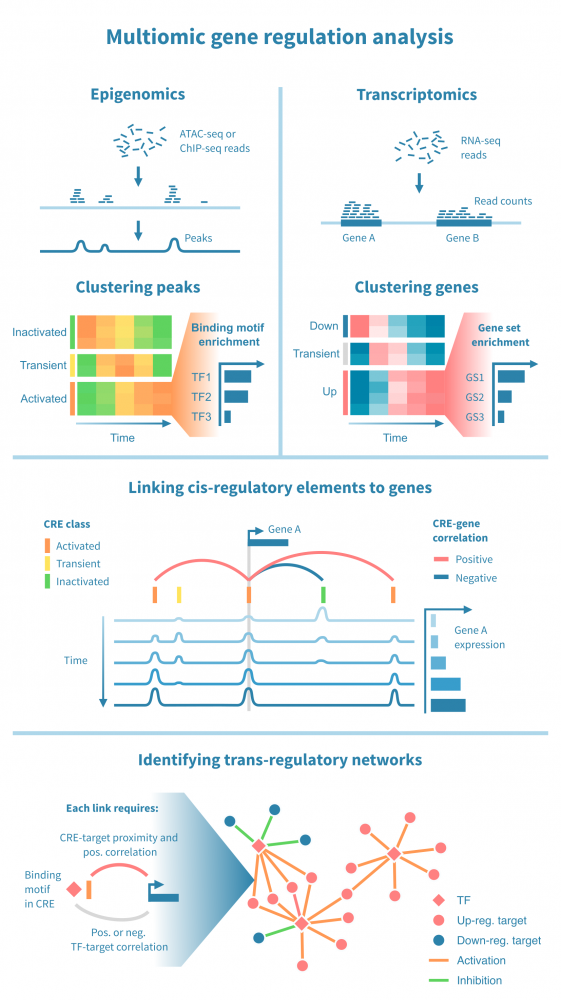
工作流程中的关键步骤包括:
- 分类顺式调节元素: 将峰值解释为 CREs,可以通过聚类分析将其按时间模式分组。这可能会产生几个 CREs 类别,其表观状态可以基于观察到的模式 (例如,激活、短暂激活、恒定活性) 和测量到的表观基因组信号 (例如,在 ATAC-seq 情况下“可访问”,在 H3K27ac ChIP-seq 情况下“活跃”) 进行分类。CRE 群集可以进一步通过富集结合基序进行注释。
- 按照它们的时间表达模式将基因分组: 同样,基因通过聚类分析分组,并基于观察到的模式 (例如,“上调”,“恒定表达”) 进行分类。基因簇通过基因集富集分析进行注释,以将它们与生物功能和过程联系起来。
- 将 CREs 与基因联系起来: 将假定的 CRE 与基因联系起来依赖于两者之间的基因组近距离关系以及 CRE 的表观活性与基因表达的相关性。
- 将 TFs 与目标基因联系起来: 建立 TF 与目标基因之间的联系依赖于 TF-CRE 链接信息 (结合基序、相关性)、CRE-目标基因链接信息 (见上文) 以及 TF-目标相关性。这种对可能的 TF-目标链接进行的多组学过滤可以识别所有这些活跃的顺式调节路径的网络。
下一步实验是什么?
上述方法描述了一个研究过程中涉及的调节程序的丰富描述。有几种方法可以进一步丰富和验证研究结果,例如:
- 通过染色体构象捕获验证CRE-靶标相互作用: Hi-C等方法可以证明基因组范围内远距离位点之间的物理相互作用。
- 通过基因组编辑验证CRE-基因相互作用: 验证调节元件在驱动基因表达方面的作用的黄金标准实验是使用CRISPR-Cas9删除CRE,并在野生型和编辑细胞中量化目标基因的表达。
- 通过ChIP-seq验证TF-CRE相互作用: 仅仅因为在显然活跃的调节元件中存在一个结合位点并不是TF-CRE相互作用的直接证据。可以使用针对感兴趣的TF的特异性抗体进行ChIP-seq(或CUT&RUN或CUT&Tag)实验,以验证因子的物理存在。
了解更多
以上我们介绍了一种整合表观基因组和基因表达数据的方法,特别是揭示cis和trans调节相互作用。了解更多关于NGS数据分析的内容: