表观基因组数据分析

揭示发育和疾病中基因调控的表观遗传机制。
表观基因组学描述了染色质状态的微小化学修饰。DNA和相关蛋白质的表观遗传变化影响基因表达并可能导致细胞状态的改变,包括疾病。
我们分析广泛的表观基因组测序数据,以深入了解细胞内分子机制并确定疾病的生物标志物。
下面我们讨论常见的表观基因组数据类型和分析,并介绍一些我们以往的表观基因组数据分析工作。
表观基因组测定
用于表观基因组分析的高通量测定方法众多,并且不断开发新的协议。最常见的表观基因组测定方法集中在DNA甲基化、DNA结合蛋白、组蛋白修饰、染色质可接近性或染色质的三维构象。
-
DNA甲基化: 基于亚硫酸盐处理的DNA的DNA甲基化测定可以以最高分辨率确定甲基化事件。这种测定使用下一代测序(全基因组或减少表示亚硫酸盐测序)或微阵列。另一种方法MeDIP测序,依赖于免疫沉淀,分辨率较低。
-
转录因子结合和组蛋白修饰: 用于确定DNA结合蛋白,如转录因子,以及组蛋白蛋白质的化学修饰的测定方法利用抗体。 ChIP-seq是最常用的方法,但已开发出具有更好分辨率的新方法。这些包括ChIP-exo、Chipmentation、CUT&RUN和CUT&Tag。
-
染色质可接近性: 映射开放染色质区域的黄金标准测定方法是ATAC-seq。ATAC-seq已经取代了先前的方法,例如DNase-seq和FAIRE-seq。
-
染色质构象: 染色质三维构象的重要性最近得到了特别的认识。染色质构象测定用于研究基因与其远端调节元素之间的物理相互作用,以及导致染色质环绕的蛋白质。Hi-C是前者的典型测定方法,而ChIA-PET可以应用于后者。
研究表观基因组对基因表达的直接影响时,通常需要在同一实验中进行RNA测序实验来补充表观基因组测量。
单细胞实验,特别是单细胞ATAC测序,越来越多地与单细胞RNA测序一起进行联合分析。这可以从同一单个细胞中获得基因表达和染色质可及性的谱。

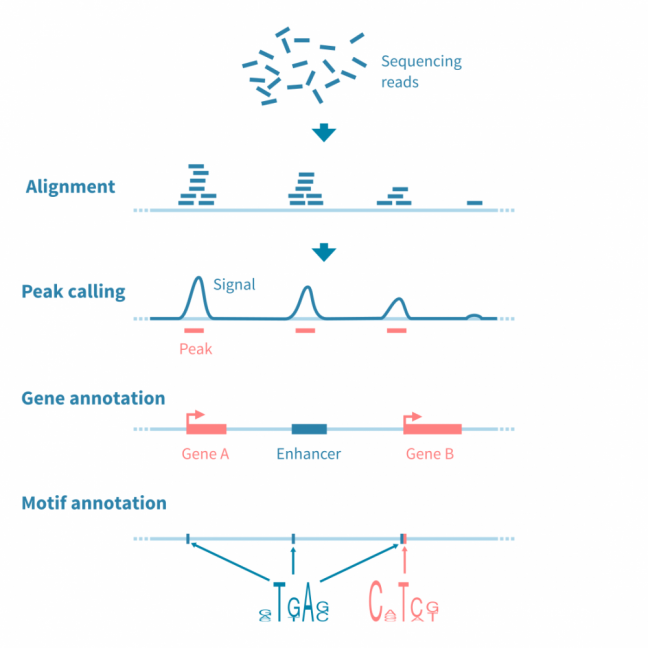
峰值calling和注释
对于大多数基于测序的表观基因组数据(特别是ChIP-seq,ATAC-seq和相关实验),分析工作流程涉及识别、注释和分析峰值,或者具有感兴趣信号的基因组区域。
首先对原始测序读数进行质量控制和参考基因组的比对,之后使用可能的对照库(在ChIP-seq的情况下,预IP输入和IP与非特异性抗体)来归一化读数覆盖信号。
使用峰值caller工具识别信号中的峰值。此阶段可能需要仔细调整参数以优化用于分析的协议。
为了进行进一步的分析,使用相关信息(如读数统计和接近或重叠的特征,如基因、调控元件和结合基序)对峰值进行注释。
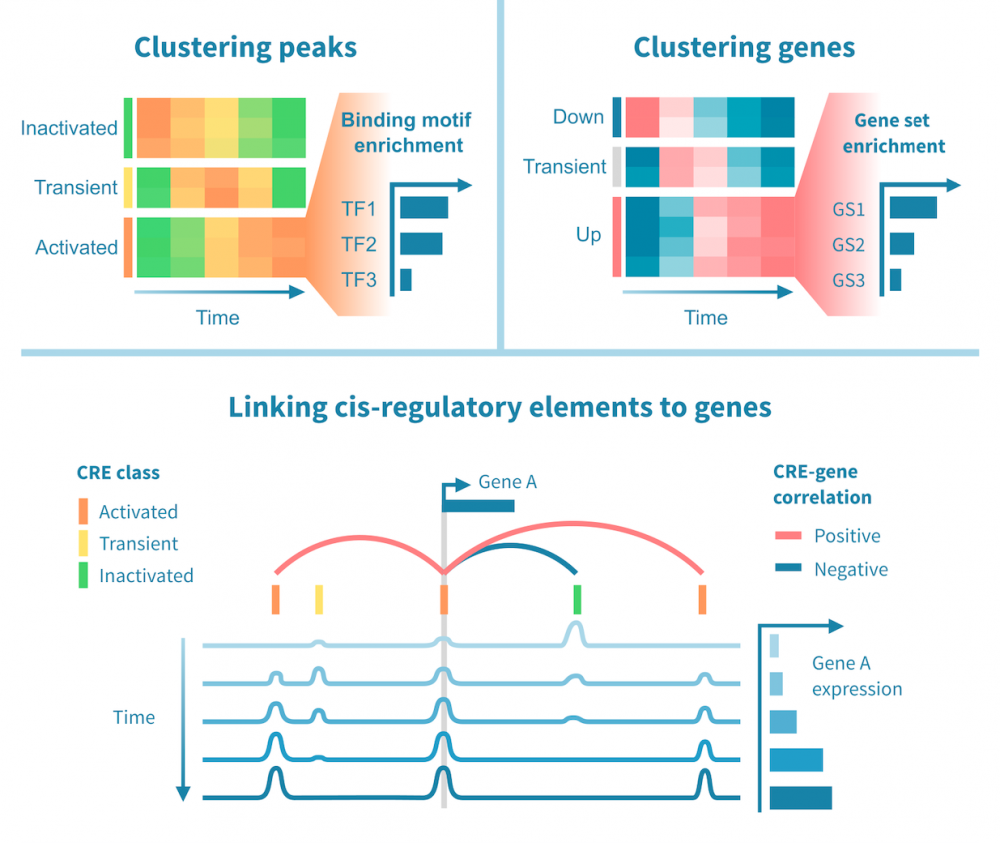
使用基因注释峰值可进行基因集富集分析,以进一步解释下游效应。
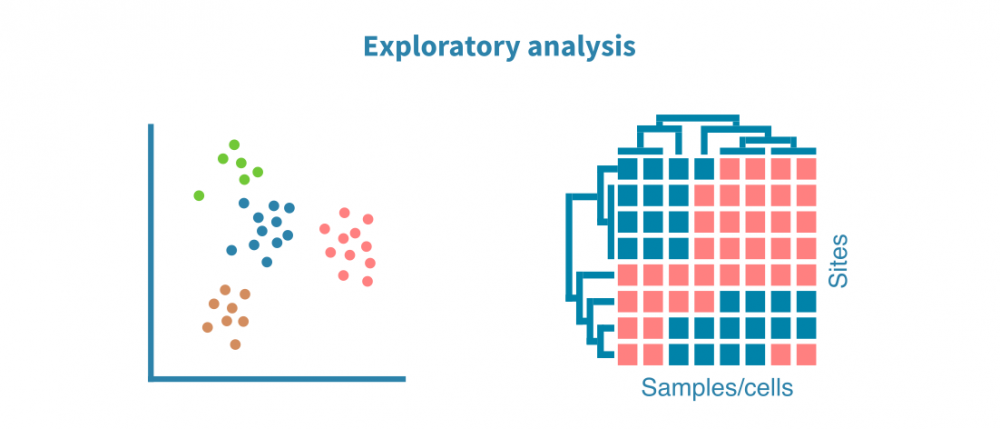
探索性分析
使用PCA(对于单细胞数据,使用UMAP或t-SNE算法)和热图可视化样本集中的注释峰值。这些可视化有助于优化峰值调用过程,并回答以下问题:
- 生物重复体在表观基因组分析方面是否相似?
- 不同的样本组(例如不同的组织、处理或时间点)是否形成单独的聚类?
- 是否存在离群样本?
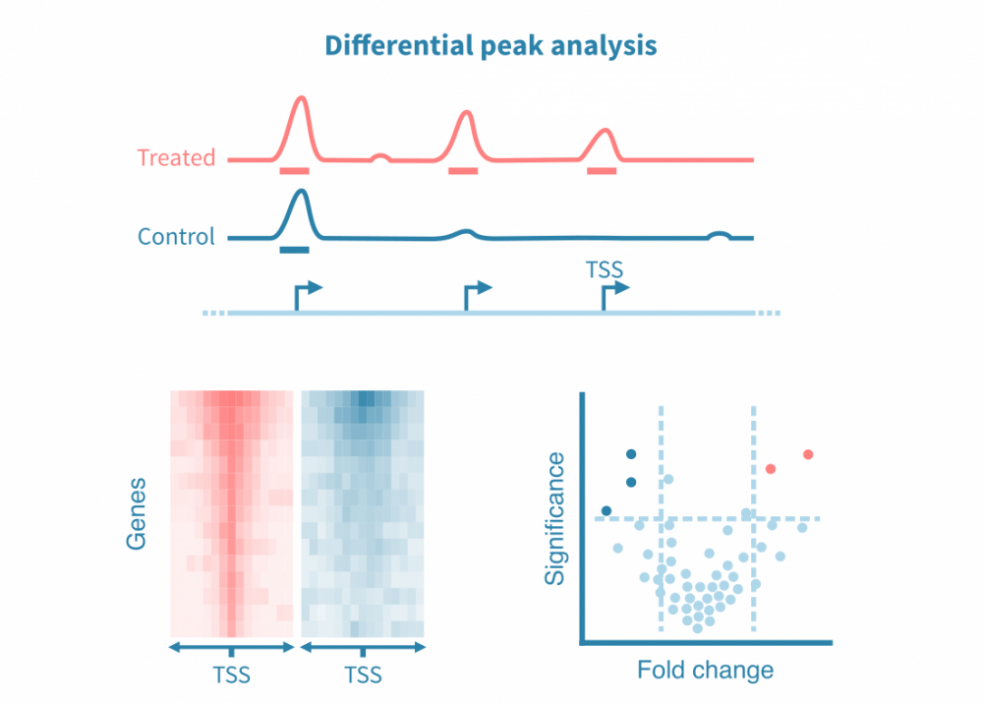
差异峰分析
为了比较不同条件,可以对已识别的峰进行统计比较,或者更常见的是直接从各自的读数覆盖信号中调用差异峰。
类似于差异基因表达分析,差异峰分析可产生效应大小和统计显着性的估计值。这些统计数据可以可视化为火山图。
由于全基因组表观基因组测量在整个基因组中产生连续的信号,因此这些分析也可以集中于特定的感兴趣区域,例如启动子或感兴趣蛋白质的已知结合位点。密度热图用于在不同条件下可视化感兴趣位点的信号。
此外,在峰值处重叠的结合基序可以在条件之间进行统计比较,并以火山图的形式进行可视化。
转录因子结合位点分析
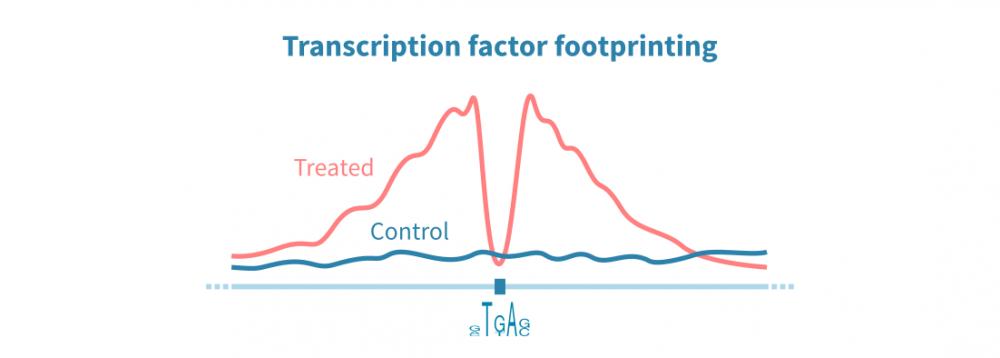
ChIP-seq和相关协议可用于在整个基因组中识别转录因子(TF)结合位点。这些检测依赖于针对感兴趣蛋白质的特异性抗体,因此这种方法可以仅识别一个TF的结合位点。另一方面,ATAC-seq数据可以通过称为TF足迹分析的方法并行识别所有DNA结合蛋白的结合位点。
在TF足迹分析中,染色质可及性信号中的窄降落被解释为蛋白质结合位点。可以间接推断TF的身份。结合RNA-seq数据,TF足迹分析可以用于以非常高通量的方式研究TF对基因表达的综合影响。
DNA甲基化数据分析
DNA甲基化数据的分析始于测序读数的质量控制和比对(或数组数据的QC和标准化),然后进行甲基化位点的调用。
检测到的甲基化位点用于识别样本之间的更大的DNA甲基化区域或差异甲基化区域(DMR)。这些区域可以类似于其他表观基因组数据中的峰值进行注释。
DNA甲基化数据的可能下游分析包括:
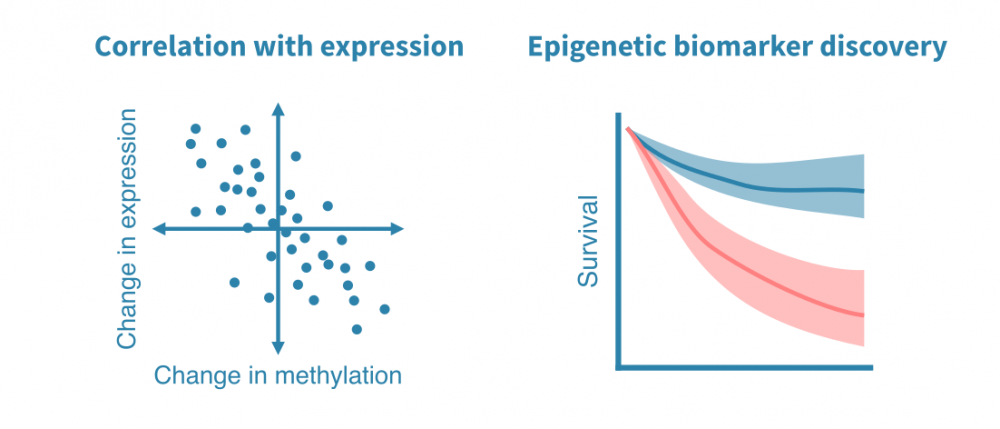
- 与基因表达数据的整合: 当来自同一环境的RNA-seq或其他基因表达数据可用时,可以研究启动子甲基化和基因表达之间的关联。
- 表观遗传标记物的发现: 来自患者样本的DNA甲基化数据可以发现临床相关的表观遗传标记物。
- 生物年龄分析: 针对DNA甲基化数据开发了生物年龄的表观模型。这些模型可用于估计个体或特定组织的生物年龄,而非年龄。
RNA-seq和表观基因组数据的整合
在同一样本上进行RNA-seq和表观基因组测序(如ChIP或ATAC-seq)可以进行综合分析,以研究基因调控程序的全基因组。
可以识别增强子和其靶基因之间的调节连接,以及转录因子和它们的靶基因,借助来自基因表达和调节元素表观基因组状态的证据建立。