DNA测序数据分析

通过DNA测序数据分析了解遗传变异和突变的影响。
DNA测序有多种形式,包括全基因组测序(WGS)、全外显子组测序(WES)和目标测序,可以研究遗传和体细胞DNA变异。除了NGS数据外,SNP和CGH阵列也可用于识别遗传多态性和拷贝数变异。微生物群落的宏基因组全基因组测序可用于分析它们的组成和功能。
我们经常分析DNA序列数据来回答基础生物学和生物医学设置中的研究问题。以下是一些典型的DNA测序数据分析。
变异分析
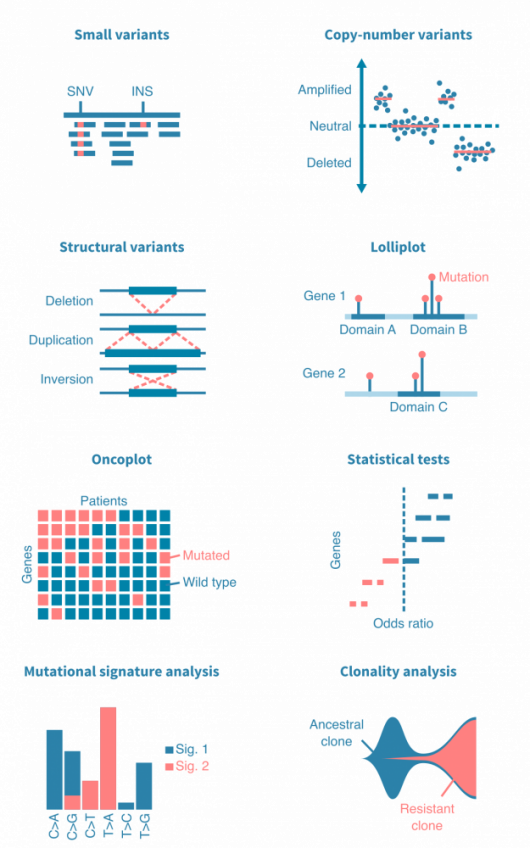
在大多数情况下,DNA测序用于识别和分析遗传变异。这些变异可以是小的核苷酸置换、插入、删除、拷贝数改变或结构变异。此外,它们可能是遗传多态性或体细胞突变。
变异分析通常从原始DNA测序数据的质量控制开始,并将测序读取与参考基因组进行比对。然后可以计算出样品与公共参考或不同样品之间不同的变异。
变异分析的关键部分是注释检测到的变异。注释,例如等位基因频率(在样本和gnomAD等公共数据库中),对蛋白质结构或基因调控的预测影响以及预测的致病性,可用于下游分析和解释中灵活选择或排名变异。
癌症研究中的变异分析通常侧重于识别加速肿瘤发生的体细胞突变(驱动突变)或可用于诊断患者或预测其疾病进程的突变。然而,非驱动突变(乘客)也携带信息。它们增加了对突变特征和癌细胞克隆性分析的可靠性。了解更多有关 癌症研究中的突变分析。
基因组组装
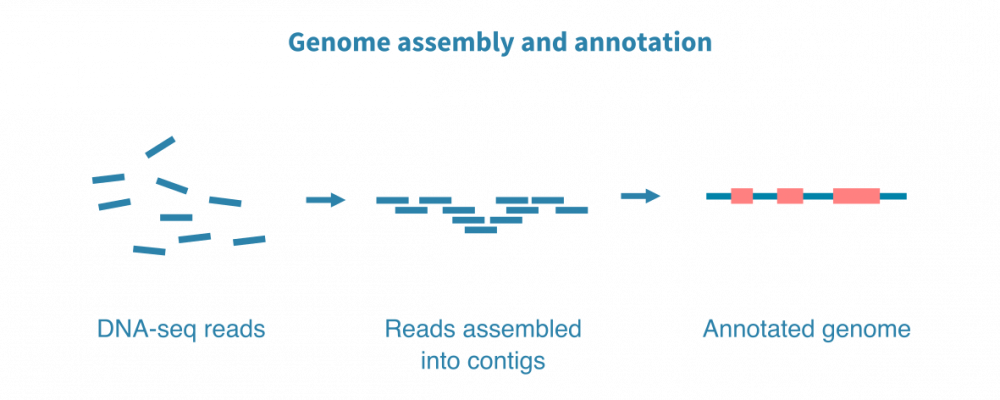
对于没有参考基因组或基因组高度动态的生物,DNA测序数据分析从组装一个全新的基因组开始。基因组组装受益于深度全基因组测序。
一个组装好的基因组会基于序列同源性、预测基因序列以及(如果有的话)来自同一生物体的RNA测序数据进行注释。如果存在近缘物种的注释基因组,可以通过将基因信息转移到新组装的基因组中来改进注释。
组装好的基因组的质量通过指标(如N50、L50以及高度保守的同源基因的完整性)进行评估。新的高质量基因组可以进行全基因组分析、群体遗传学等等!
宏基因组学
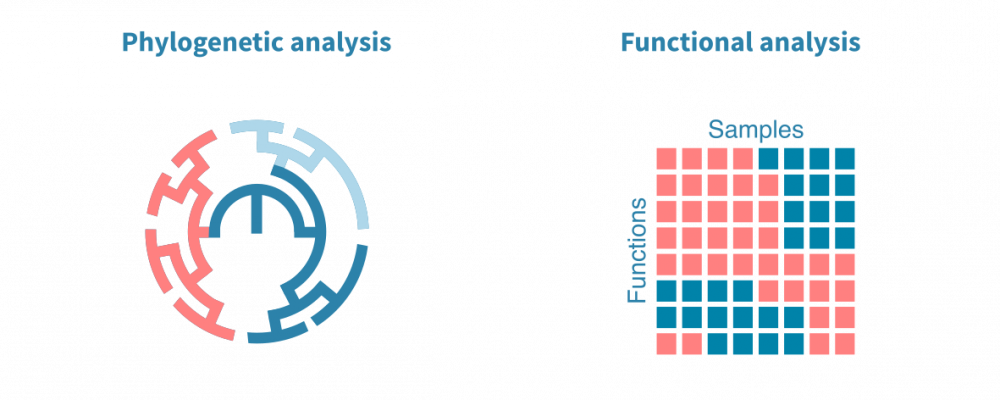
宏基因组学提供了对生态位中微生物多样性的无偏视图,包括来自寄主生物体和土壤的样品。使用shot-gun全基因组测序数据,reads被组装成contigs并分配给物种或操作分类单元(OTUs)。
已确定的物种或OTUs被组织成系统发育并进行定量。通过使用公共数据库,可以确定序列社群中单个基因或多基因途径所带来的功能。
请注意,16S引物子测序是宏基因组测序的一种经济实惠的替代方法,可用于识别物种并构建系统发育,但不允许进行高质量的功能分析。
群体遗传学
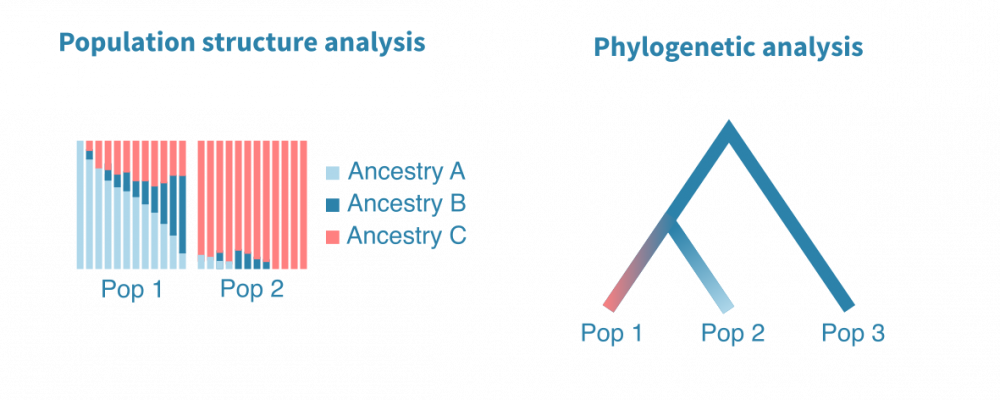
从相关种群中采样的个体的全基因组测量包含有关群体结构、谱系和历史的丰富信息。非模式生物的群体遗传分析通常从基因组组装和注释开始,然后进一步确定样本群体中的遗传多态性。基于这些多态性及其等位基因频率的下游分析有助于研究物种形成和适应等进化现象。
典型的分析包括主成分分析、对群体内和群体间的遗传变异进行分析以识别受进化选择影响的位点,以及对群体混合、系统发育和人口历史的分析。
全基因组关联分析
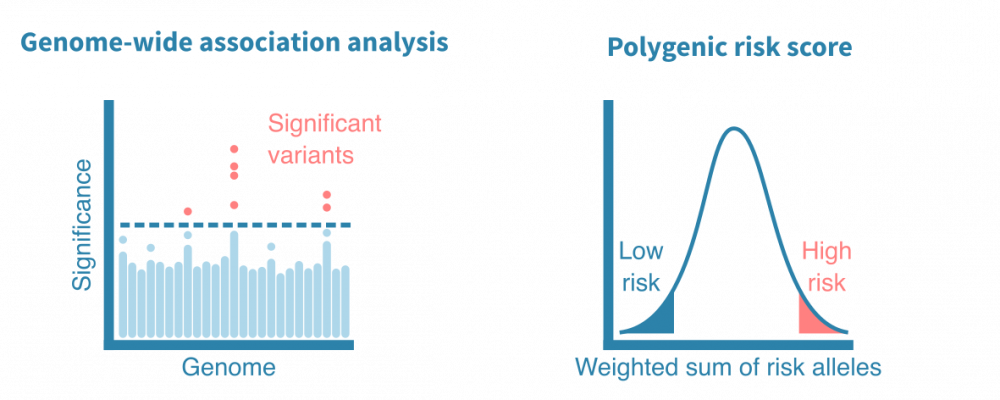
生物医学上的群体规模遗传分析旨在确定与相关表型或疾病相关的基因和变异。除了一些单基因遗传性很强的疾病外,大多数疾病需要大的群体级别样本量才能获得足够的统计力量以发现关联。这样的全基因组关联研究(GWAS)基于来自生物库或其他大型存储库的SNP阵列或DNA测序数据。
GWAS的结果会给出每个个体变异与研究疾病之间的关联的统计数据。对于多基因疾病,即使疾病具有很强的遗传性,单个变异也可能具有非常弱的效应大小。在这种情况下,可以使用多基因风险评分(PRS)来总结大量变异的效应,得出一个综合风险评分,具有潜在的临床应用。
了解更多
了解更多关于NGS数据分析的方法: